All enhancements become part of the product. No back applying of enhancements are required as part of an upgrade. Upgrades are included as part of maintenance.
Upgrades Included As A Part Of Maintenance
All Innovatum customers who are current with their maintenance are presented with the option of upgrading to newer versions with additional functionality at no extra charge for the newer software version.
All In One Solution
That includes data maintenance, labeling and regulatory compliance.
We offer
Best Support In Industry
Unmatched issue resolution times and customer satisfaction ratings
Adobe Suite Artwork Management
Versions & manages native Adobe files- associates them with items
Unmatched Risk Reduction
Commonly referred to as a quality management system for labeling
True Enterprise Collaboration
Browser based with unlimited access licensing for your complete supply chain
Compliance Capability Built-In
Completely 21 CFR and Annex 11 capable with reporting
Manages Data and Regulatory Submissions
GUDID, EUDAMED, GS1 Data Pool, Others
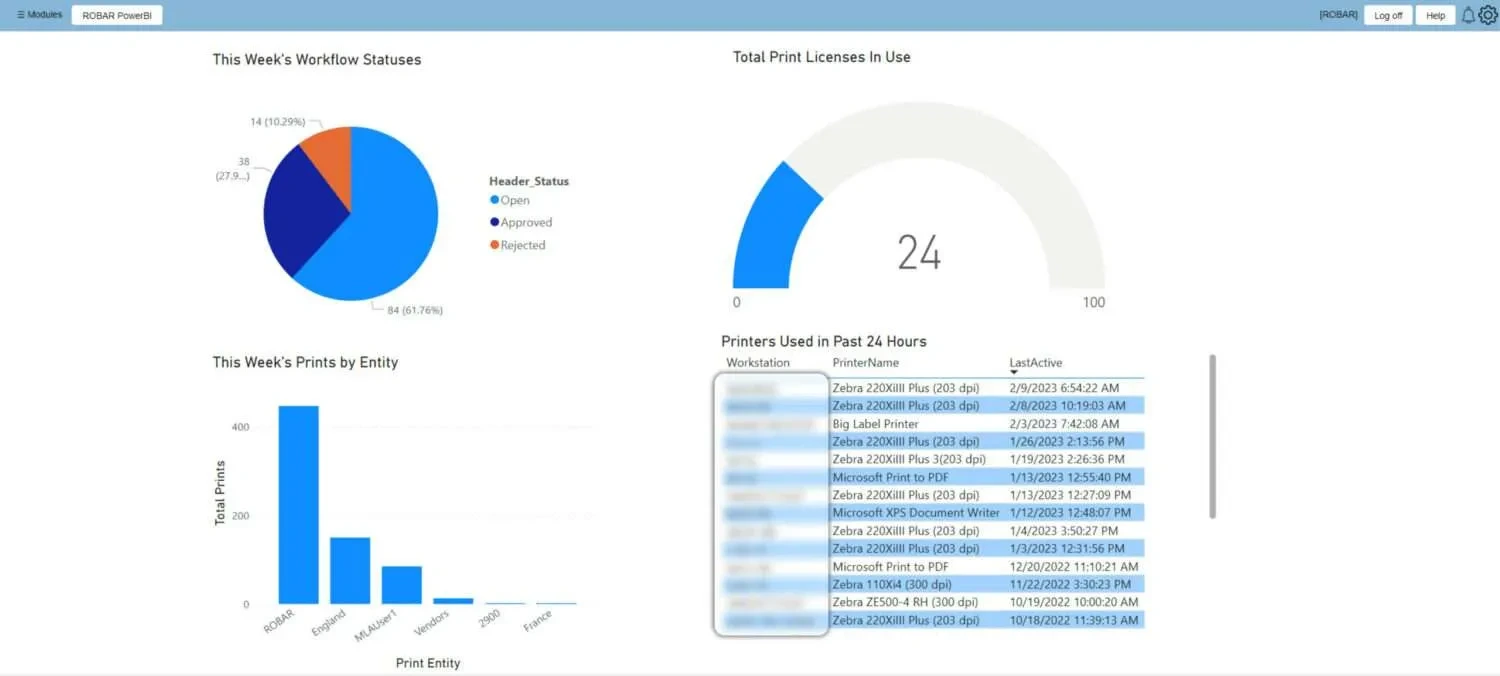
System Dashboard
We Work With Organizations Around the World
We provide complete systems, modular systems, consulting, training, and exemplary 24/7 support services across all borders and time zones.
Innovatum’s ROBAR provides many powerful capabilities for UDI/MDR/IVDR/Eudamed and is delivered with consulting, implementation, and validation assistance. As a true end-to-end regulated labeling system provider, Innovatum has been a top innovator in life sciences labeling for over 25 years. Innovatum’s fully configurable and easily validatable RIMS/PIMS/MDM labeling systems are easily expandable to meet future regulatory needs without involving the IT department. Additional modular capabilities include 100% label inspection and elFU management with hosting.